
Biological processes are regulated by complex networks of interacting messages called signalling pathways. To occur at rates fast enough to sustain life, these biological processes need enzymes – biological catalysts that speed up chemical reactions. One important family of enzymes crucial for regulating cell function is the protein kinase C (PKC) family. PKCs have been shown to be involved in a diverse range of biological processes including the regulation of cell growth, differentiation, metabolism, and cell death (apoptosis). As well as their role in normal biological processes, PKCs have been implicated in maladaptive pathological responses that drive a wide range of clinical disorders including diabetes, Alzheimer’s disease and cardiovascular disorders. Consequently, the PKC family has been the focus of intensive research ever since its discovery in 1977.
Professor Steinberg has revolutionised the thinking behind the intricate roles of PKCδ in health and disease.
One enzyme, many forms
PKC was originally thought to be a single protein. We now understand that PKCs comprise of a family of structurally related subtypes (called isoforms); they are classified depending on their structure, function, and cofactor (helper molecule) requirements. PKCs are serine/threonine kinases; they exert intricate control over various aspects of protein function in health and disease by transferring a phosphate to the hydroxyl (OH) group of either a serine or threonine residue in an acceptor protein. This process is called phosphorylation.
One particular PKC isoform – protein kinase C-delta (PKCδ) – is the focus of research for Professor Susan Steinberg, Professor of Pharmacology at Columbia University Irving Medical Center. An important enzyme, PKCδ has the ability to phosphorylate multiple target proteins involved in a diverse range of biological processes in the heart, both in health and disease, including regulation of cardiac muscle contraction, the severity of ischemia/reperfusion injury, and the pathogenesis of cardiac hypertrophy and failure. Elegantly combining a wide range of reductionist molecular and cell biological approaches, Professor Steinberg has revolutionised the thinking behind the intricate roles of PKCδ in health and disease. Her ultimate aim is to develop potential therapies targeted to PKCδ-dependent pathological cardiac remodelling.
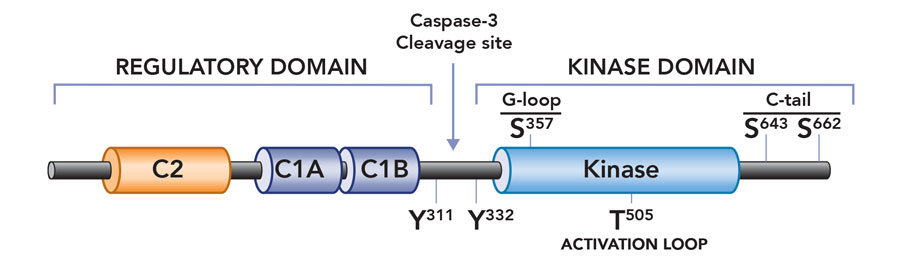
Context-specific roles of PKCδ
Professor Steinberg’s early interest in PKCδ was sparked by intriguing differences that set it apart from other enzymes in the PKC family. The hallmark of activation for a conventional PKC is its translocation to the cell’s plasma membrane. After docking to the membrane, PKC sits near target substrates where it carries out its catalytic effects. Professor Steinberg noted that this mechanism failed to explain PKCδ’s actions in heart cells, where PKCδ phosphorylates proteins away from the cell membrane. Professor Steinberg also observed that PKCδ has diverse and in some cases opposing actions – for example its role in both ischemic/reperfusion injury and cardioprotection. This did not fit with the idea that PKCδ’s catalytic ability is an inherent property of the enzyme, which remains unaltered when the enzyme is activated.
To address this quandary Prof Steinberg embarked on a series of careful experiments in heart cells (cardiomyocytes). She provided compelling evidence that PKCδ is activated in a stimulus-specific manner in cardiomyocytes. Specifically, she showed that growth factor stimuli activate PKCδ at lipid membranes, but PKCδ becomes a lipid-independent enzyme during oxidative stress. Importantly, this change means it is poised to phosphorylate substrates throughout the cell. An additional observation suggested a molecular mechanism to explain this change in enzyme function: that oxidative stress results in the activation of Src family tyrosine kinases (a family of enzymes that phosphorylate target proteins on tyrosine residues), and that PKCδ is unique among PKC family enzymes in that it is a target for regulatory tyrosine phosphorylation by Src kinases. In a series of carefully designed experiments, Professor Steinberg used molecular and biochemical approaches to show that oxidative stress leads to an increase in PKCδ phosphorylation at tyrosine-311, a tyrosine residue in the hinge region of the enzyme that is the major site for PKCδ phosphorylation by Src. She then showed that phosphorylation at this tyrosine residue converts PKCδ from a lipid-dependent serine kinase (an enzyme that acts primarily to phosphorylate target substrates on serine phosphoacceptor sites) to a lipid-independent serine/threonine kinase (an enzyme that can now phosphorylate substrates on either serine or threonine residues). This redox-dependent acquisition of a new enzyme activity (i.e., the ability to phosphorylate an entirely new set of substrates with threonine residues at their phosphoacceptor sites) would explain the oxidative stress-dependent changes in PKCδ’s cellular actions. Professor Steinberg is now using genetically modified mouse models to explore the role of the oxidative stress-induced acquisition of new PKCδ enzymatic activities in the pathogenesis of cardiac ischemia-reperfusion injury.

Fine-tuning PKCδ activity
Subsequent mutagenesis studies addressed the dilemma of how a phosphorylation at tyrosine-311 (in the hinge region of the enzyme, at some distance from the active site or ‘catalytic pocket’ of the enzyme) alters enzyme activity. Taking advantage of mass spectrometry techniques, Professor Steinberg identified a novel phosphorylation site at serine-357 in the catalytic pocket of PKCδ – structural models suggest that this serine residue points to the phosphoacceptor site on a target substrate. Professor Steinberg then showed that PKCδ is recovered with a high level of serine-357 phosphorylation at baseline, and that oxidative stress and the resultant tyrosine-311 phosphorylation leads to a major conformational change in the enzyme that results in a decrease in phosphorylation at serine-357 (presumably because this residue becomes exposed and susceptible to dephosphorylation by cellular phosphatases). Importantly, molecular and biochemical studies then established the critical role of this serine-357 phosphorylation site, showing that the decrease in serine-357 phosphorylation is sufficient to convert PKCδ from a lipid-dependent serine kinase to a lipid-independent serine/threonine kinase. These studies implicated serine-357 phosphorylation as a novel dynamically-regulated post-translational modification – in essence, a switch that mediates the oxidative stress-induced increase in PKCδ’s lipid-independent kinase activity and change in substrate-specificity. This mechanism is novel not only for PKCδ but unprecedented for any other kinase.

PKCδ is converted into a constitutively active catalytic domain fragment as a result of caspase-3 cleavage during apoptosis.
Steinberg continues to unravel the agonist-specific PKCδ signalling ‘modes’ that link the enzyme to functionally distinct cellular responses.
New insights into PKCδ
The diverse roles of PKCδ demonstrate the central importance of this enzyme in biological processes. Building on her ground-breaking work, Professor Steinberg continues to unravel the different agonist-specific PKCδ signalling ‘modes’ that link the enzyme to functionally distinct cellular responses. Her future work will use genetically modified mouse models to define the functional consequences of the oxidative stress-dependent changes in PKCδ activity, with a specific focus on the role of PKCδ in cardiac contraction and ventricular remodelling. Importantly, she aims to identify potential targets for the design of inhibitors that are tailored to prevent PKCδ-driven mechanisms that contribute to pathologic cardiac remodelling.
There have been several ‘Ah-Ha’ moments in my career, particularly as it relates to studies of PKCδ. The satisfaction of conjuring up a unifying/simple model to explain previously confusing experimental data and then being able to guide the many talented members of my laboratory as they painstakingly pursue various experimental strategies to ultimately substantiate our initial hypothesis cannot be overstated. Similarly, the ability to use biochemical approaches to identify novel molecular determinants of PKCδ – in essence using a biochemical strategy to infer structural information typically defined using crystallographic methods that directly interrogate protein structure – also has been extremely rewarding.
References
- Distinctive activation mechanisms and functions for protein kinase Cd. Steinberg SF. Biochem J 2004; 384: 449-459.
- Mechanisms for redox-regulation of protein kinase C. Steinberg SF. Front Pharmacol. 2015 Jun 23;6:128. Review.
- Cleavage alters the molecular determinants of protein kinase C-δ catalytic activity. Gong J, Park M, Steinberg SF. Mol Cell Biol. 2017 Aug 7. pii: MCB.00324-17.
- Protein kinase C mechanisms that contribute to cardiac remodelling. Newton AC, Antal CE, Steinberg SF. Clin Sci (Lond). 2016 Sep 1;130(17):1499-510.
- The C2 domain and altered ATP-binding loop phosphorylation at Ser359 mediate the redox-dependent increase in protein kinase C-delta activity. Gong J, Yao Y, Zhang P, Udayasuryan B, Komissarova EV, Chen J, Sivaramakrishnan S, Van Eyk JE, Steinberg SF. Mol Cell Biol. 2015 May;35(10):1727-40.
Professor Steinberg’s research focuses on the signalling enzyme, protein kinase C-delta (PKCδ).
Funding
NIH – HL123061
Collaborators
- Dr Jennifer Van Eyk
- Dr Ju Chen
- Dr Sivaraj (Shiv) Sivaramakrishnan
Bio
 Dr Steinberg earned a BS in Biology from MIT and an MD from Harvard Medical School. She is currently Professor and Director of Graduate Studies in Pharmacology at Columbia University, a member of the American Heart Association Basic Science Council Leadership Committee and a Fellow of the International Society for Heart Research.
Dr Steinberg earned a BS in Biology from MIT and an MD from Harvard Medical School. She is currently Professor and Director of Graduate Studies in Pharmacology at Columbia University, a member of the American Heart Association Basic Science Council Leadership Committee and a Fellow of the International Society for Heart Research.
Contact
Prof Susan F. Steinberg
Director of Graduate Studies, Department of Pharmacology
Professor of Pharmacology
Columbia University
P&S 7-443
630 West 168 Street
New York, NY 10032
USA
E: [email protected]
T: +1 212-305-4297
W: www.cumc.columbia.edu/dept/gsas/pharm/steinberg.html
Creative Commons Licence
(CC BY-NC-ND 4.0) This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. Creative Commons License
What does this mean?
Share: You can copy and redistribute the material in any medium or format







