
Catalysts speed up chemical reactions by lowering the energy required for the reaction to occur. To achieve this, the catalyst forms an intermediate complex with the chemical reactants, to provide a lower energy route to reaction. These intermediate complexes are typically short-lived as, to allow the catalytic cycle to continue, the catalyst needs to be ‘refreshed’ so it can be used in future reactions.
There are many types of catalysts made up of a whole variety of different chemical elements. Some of the most commonly used elements are transition metals because they can interact with a wide variety of chemical species. These elements can also be arranged in a variety of ways to make catalysts with different shapes and structures, that can have a dramatic effect on their efficiency and properties.
Johnston and Baletto aim to tune catalysis at the nanoscale using state-of-the-art numerical and computational techniques![]()
As catalysts play a key role in processes such as the removal of toxic carbon monoxide from exhaust fumes, there is a huge need to develop new catalytic species that are efficient, robust and cost-effective. However, intelligently designing new catalysts or predicting which materials are likely to be successful in the lab relies on a deep understanding of the intricacies of catalytic mechanisms, another very challenging problem in itself. Prof Roy L. Johnston and Dr Francesca Baletto at the University of Birmingham and King’s College London, with their collaborators, are developing and applying computational techniques for exactly this problem, via the design and tailoring of nanomaterials for catalysis.
Catalysis bit by bit
Modelling catalytic materials computationally is no mean feat. While computational modelling can be used to predict the outcomes of simple gas phase chemical reactions very accurately, modelling solids, and particularly those with heavy atoms like metals, offers a number of additional challenges. Prof Johnston and Dr Baletto are also particularly interested in the properties of more unusual materials, known as nanoalloys, which have numerous desirable and enhanced catalytic properties. These nanoalloys are nanoparticles which are made up of two or more metallic elements.
Computational modelling of a catalytic reaction requires an understanding of the structure of the catalyst itself as well as any substrates it interacts with. The nanoalloys that Prof Johnston and Dr Baletto study are particularly complex as they come in a variety of sizes. This means that the computational models need to trial a huge number of different configurations and particle sizes to explore which combination of structural features will give rise to the most promising catalysts.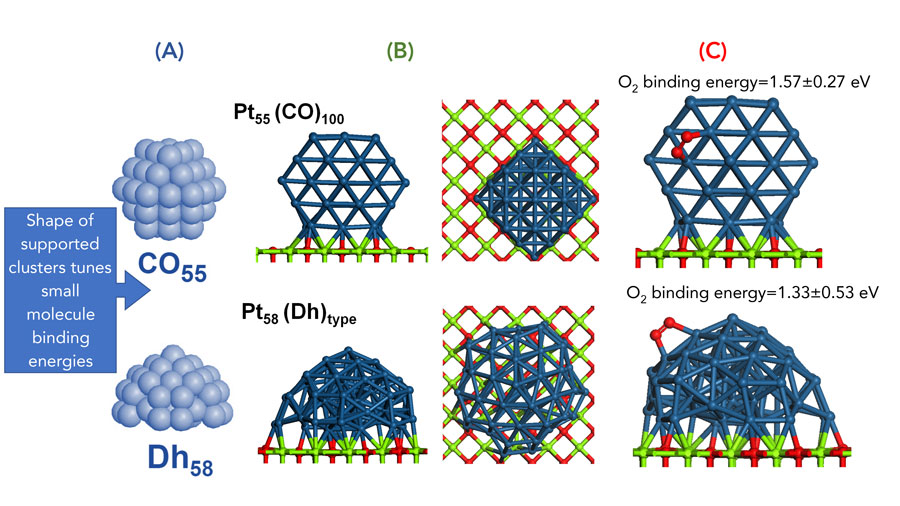
Johnston and Baletto are searching for descriptors to relate the structures of metallic nanoparticles to their catalytic performance![]()
Even greater levels of complexity are introduced into the computational models by potential transformations of the structure of the catalyst during a reaction event. While such changes in the shape of the catalyst may be rare events, it is necessary to account for these possibilities in the computational modelling to accurately reproduce experiments. As well as the development of new catalytic materials, this is also driving the development of new computational methods, as part of the TOUCAN project (‘Towards an Understanding of Catalysis on Nanoalloys’), a collaborative network of projects focused on understanding how nanoparticles and nanoalloys can speed up chemical reactions.
Greener energy
One of the key reasons that Prof Johnston and Dr Baletto are so interested in nanoalloys over traditional, substrate-supported monometallic catalysts is the potential impact they could have in the field of sustainable energy and clean technology. They have studied the impact of nanoalloys in the catalysis of a variety of reactions, including the oxygen reduction reaction (ORR), which is of importance in electrochemical fuel cells, and the oxidation of carbon monoxide (CO). As well as being a toxic pollutant, CO is one of the main poisons of fuel cell catalysts; it decreases the efficiency of the platinum electrodes, which are incredibly costly to replace due to the increasing scarcity of platinum.
Unfortunately, there are many existing catalysts for which platinum is an essential component and it has been challenging to find chemical elements that can act as a suitable replacement. However, platinum nanoalloys offer a route to making catalysts that are as efficient, but require much less platinum by replacing some of the platinum content with a cheaper metal. Prof Johnston and Dr Baletto have been modelling the effects of the amount of substitution in the nanoalloys, for a range of different metals, to see if they can identify which metals, and how much of each one, are required to design novel, highly efficient and selective catalysts for reactions such as CO2 capture and reduction, and O2 reduction.
The work of Prof Johnston and Dr Baletto is helping to provide an unparalleled insight into exactly how catalysts work, as well as leading the development of new, general computational approaches, that can be used to predict the properties of a variety of catalytic species. All of this is helping to drive ‘intelligent’ catalyst design. This will help identify and pre-screen species that are likely to be successful candidates, essential not just as a cost-saving exercise but to accelerate the development of a new generation of catalysts in clean technologies.
- Developing cheaper catalysts, by replacing or reducing scarce metals such as Pt and other precious metals.
- Knowledge of the atomistic mechanisms, including reaction kinetics and intermediate formation and the effect of the surrounding environment. This will allow the correlation of nanoshapes with catalytic properties, leading to new design rules.
- Modelling nanocatalysis as a dynamic process occurring at high temperatures and pressures, in the presence of various reactants and products. This will be facilitated by the development of new theoretical methods which are able to capture and analyse various elementary steps including the structural, and hence electronic, evolution of the nanosystems.
Do you think nanocatalysts will be the dominant type of catalyst in the future?
Yes, since nanoparticles allow the tailoring of catalytic properties by changing the elemental composition and ordering, shape and morphology, which in turn tunes the electronic properties of the nanoparticle. The global market for nanocatalysts is continually expanding and this trend is forecast to continue for the near and medium-term future. Metallic, multimetallic (mixing precious and abundant metals) and hybrid (e.g., metal-metal oxide and metal-biomolecule) nanoparticles will play an increasingly important role, once the manufacture of highly efficient catalysts, controllable at the molecular scale, becomes feasible at a reasonable cost. The combination of numerical and experimental tools is mandatory to achieve the required control.
What do you think are some of the most exciting developments in catalysis that have arisen from using computational modelling in this area?
The most exciting idea is the growing idea that chemistry can be tuned by modifying nanoparticle shape, leading to a completely new approach to catalysis which is only feasible at the nanoscale. In this regard, a hot topic is the search for descriptors, which are measurable quantities that link nanoparticle shape and chemical composition to their catalytic activity and selectivity. Finding robust descriptors will open the possibility of developing a dynamical model for nanocatalysis, where the substrate is also mobile. Numerical tools have begun to be used to elucidate the role of the surrounding environment, at least as far as it stabilises particular shapes and chemical ordering of nanoparticles
What are the main challenges to be overcome for computational methods to achieve ‘chemical accuracy’ in describing catalytic processes?
- To include all aspects of the environment of the catalyst, including the support (usually a metal oxide material), temperature, pressure, solvent (for solution phase catalysis) and any surface-bound but non-reactive ligand species.
- Atomistic methods in combination with machine learning tools can achieve the desired accuracy needed to shape nanocatalysts with the most promising characteristics, based on the use of descriptors which will define proper design rules.
- As density functional theory is at the core of catalysis modelling, the choice of pseudo-potentials with the right level of accuracy is important, with an eye to size scalability.
- A dynamical model for understanding chemical reactions in a realistic environment, including atomic mobility within each nanoparticle.
- New numerical tools are needed to understand the assembly of nanoparticles into super-architectures and the influence this can have on catalysis.
Do you think it will be possible to deduce general intuitive chemical models for catalyst design from your work?
Yes, the search for and definition of geometrical descriptors in conjunction with experimental data – e.g., electron microscopy – will open a new way to design nanocatalysts in an intuitive fashion. The use of machine learning is also likely to shed new insights on how catalytic activity depends on nanoparticle shape, size and chemical ordering, leading to new and fascinating routes for nanocatalyst design. Existing optimisation tools can be adapted to explore the pool of candidates which maximise/minimise the geometrical quantities of interest, eventually providing guidelines for the synthesis of these nanoparticles.
Prof Johnston and Dr Baletto use computational techniques to design and tailor nanomaterials, focusing on nanoalloys and their catalytic properties.
Funding
EPSRC (Critical Mass Grant EP/J010804/1 and EP/J010812/1)
Collaborators
Other PIs on the TOUCAN Project:
Prof Gábor Csányi, Prof Chris Pickard, Prof David Wales (University of Cambridge); Dr Jonathan Doye (University of Oxford)
External Collaborators:
Professor Dr Rolf Schaefer (Technical University Darmstadt, Germany); Prof Micha Polak (Ben Gurion University of the Negev, Beer Sheba, Israel); Prof Marcela Beltran (UNAM, Mexico); Dr Laurent Piccolo (CNRS, Lyon, France); Prof Roberto D’Agosta (ETSF-Nanobio, EHU/UPV, San Sebastian, Spain); Dr Caetano Miranda (USP, Sao Paolo, Brazil); Dr L. Oliver Paz-Borbon (UNAM, Mexico)
Bio
 Roy Johnston is Professor of Computational Chemistry at the University of Birmingham, UK. He has published approximately 250 journal articles, reviews, books and book chapters.
Roy Johnston is Professor of Computational Chemistry at the University of Birmingham, UK. He has published approximately 250 journal articles, reviews, books and book chapters. Francesca Baletto is a Senior Lecturer in Physics at King’s College London, UK. She is authors of more than 45 journal articles, reviews, and book chapters.
Francesca Baletto is a Senior Lecturer in Physics at King’s College London, UK. She is authors of more than 45 journal articles, reviews, and book chapters.Contact
Professor Roy Johnston
School of Chemistry
University of Birmingham
Edgbaston,Birmingham
B15 2TT, UK
E: r.l.johnston@bham.ac.uk
T: +44 121 414 7477
Dr Francesca Baletto
Physics Department
King’s College London
WC2R 2LS, UK
E: francesca.baletto@kcl.ac.uk
T: +44 2078482152
More info:








