
- Indigenous Americans of southwestern USA have one of the highest rates of Type 2 diabetes (T2D) in the world.
- Robert C Williams, Emeritus Professor and NIH Special Volunteer, and colleagues perform in-depth genetic analysis to unmask the roles of the Human Leukocyte Antigen (HLA) gene and the SLC16A11 gene in the development of T2D in this population.
- Epistasis, a genetic phenomenon, exists between the protective HLA-DRB1*16:02 gene and the ancient mutation at-risk gene, SLC16A11, so that its combination can give protection to up to half of those carrying it.
- Surprisingly, this gene combination puts leaner individuals at risk and protects those with a higher body mass index (BMI) – a puzzling finding that needs exploring.
Thousands of genes in our cells code for proteins that orchestrate an array of biological functions. Their complex interplay along with environmental factors determine health and disease. Advanced genotyping technologies enable extensive genome sequencing – this means that the combination of DNA pairs making up genetic information can be analysed to better understand the pathology of disease.

Using such techniques, including computer algorithms, are researchers in the genetics section of the NIH Intramural Branch in Phoenix, Arizona whose work focuses on human genetic variations and the risk of Type 2 diabetes (T2D) in Indigenous Americans. Professor Robert C Williams and his team conducted a recent study which suggests a genetic phenomenon between two genes that confers protection against T2D in overweight, not lean individuals. Such unexpected, intriguing results warrant further investigation.
A gene with many faces
The Human Leukocyte Antigen is a system of genes that code for proteins essential to immune system functioning. We inherit these genes, one group (haplotype) from each parent. More specifically, HLA proteins present foreign peptides (the fragments that make up the building blocks of proteins) to T cells of the immune system to kickstart an immune response. HLA is famously polymorphic, meaning it comes in many different forms or alleles so that all bases are covered for recognising foreign antigens and initiating an immune response. Two main classes of HLA exist, class I and II, which are further subtyped into alleles (for example HLA-A, HLA-DR) and within these are variants usually denoted by an asterisk and numbers (for example HLA-DRB1*16:02).
Even though the complex HLA system helps recognise foreign pathogens for elimination, in autoimmune diseases this system goes wrong and battles to distinguish between our own proteins and foreign ones. One such autoimmune disease is Type 1 diabetes (T1D) where our own immune system destroys pancreatic cells that produce insulin.
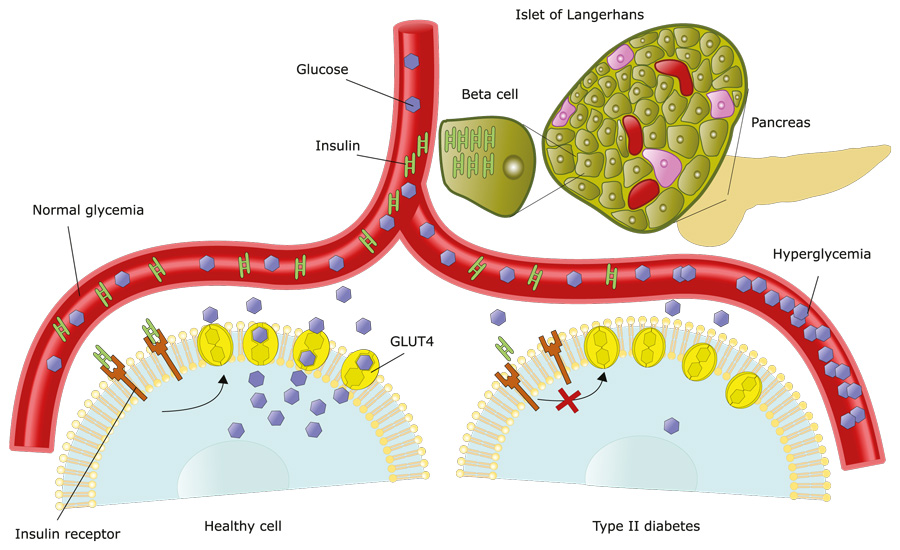
The HLA is a known genetic determinant of this type of diabetes, but until more recently it had not been implicated in T2D. With HLA alleles a risk factor for T1D pathology, Williams and his team suspected these genes may also play a role in T2D and undertook extensive genotyping in an Indigenous American population to investigate this.
Protection
Marked by obesity, hyperinsulinemia (a high amount of insulin in the blood), increased insulin resistance, and too much internal glucose production, T2D is highly prevalent in Indigenous Americans in southwestern USA. This population has one of highest T2D incidence in the world and has been studied in a longitudinal clinical and genomic study since 1965. A gene of focus is HLA-DRB1, whose job it is to present peptides to the immune T cells. When proteins are replaced in the body, they are broken down into smaller fragments called peptides and their constituent amino acids. HLA-DRB1 can ‘hold peptides’ that are 15–20 amino acids long and present them to T cells, and this kickstarts an immune response and the production of antibodies.
In 2011, the researchers identified a specific single nucleotide polymorphism (SNP) in the HLA gene that affected the expression of HLA-DRB1 (an immune-related gene) and its variant *16:02 reducing the risk of T2D. The team believe this reduced risk is because of improved immune self-tolerance conferred by HLA-DRB1*16:02. Self-tolerance is the immune system’s ability to know its own ‘self’ proteins and clearly distinguish these from foreign ones. So, the presence of HLA-DRB1*16:02 is protective against T2D and those without it secrete less insulin, presumably because of autoimmune ‘attacks’ on insulin-secreting cells. However, more research is needed to fully understand the mechanisms involved.
Protection and risk
With their 2011 paper implicating the immune system in the development of T2D, Williams and colleagues questioned whether this gene interacted with SLC16A11, a non-immune gene and known risk factor for T2D in individuals with Indigenous American ancestry. The gene SLC16A11 comes in two forms: a wildtype form and an ancient mutation form that is believed to be 750,000 years old. With one protective gene (HLA-DRB1*16:02) and one at-risk gene (SLC16A11), the team wondered what the combined effect is – do the genes act together or against each other?
The study of over 6,000 Indigenous Americans revealed that the interaction between a variant of HLA-DRB1 and one copy of the ancient SLC16A11 mutation reduces the risk of T2D in Indigenous Americans by half.
In their most recent paper, published in the journal Diabetes in 2024, the researchers answered this question by uncovering a genetic phenomenon called epistasis. In epistasis, variants from two different genes act together to affect the phenotype. The study of over 6,000 Indigenous Americans revealed that the interaction between HLA-DRB1*16:02 and one copy of the ancient SLC16A11 mutation reduces the risk of T2D in Indigenous Americans by half. Williams and colleagues wanted to take this a step further and understand peptide binding by identifying the core peptides of the ancient mutation responsible for this. Using computer algorithms, they were able to discover nine amino acids that are key to its functionality. Using the amino acid alphabet, this peptide is FSAFASGLL. The letter G is the mutation.
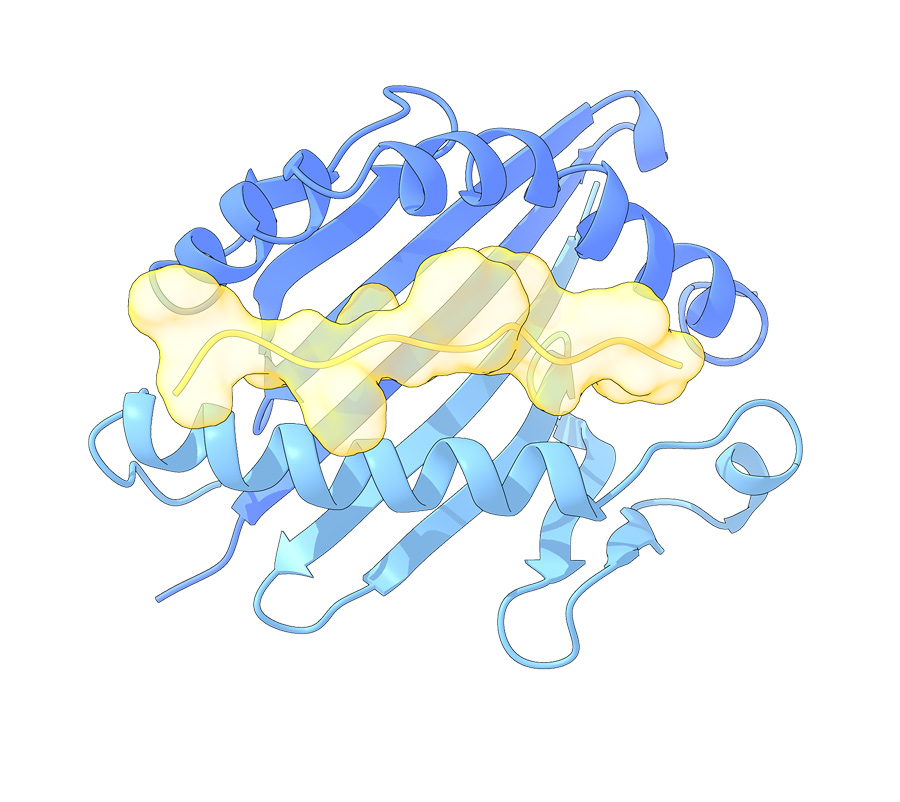
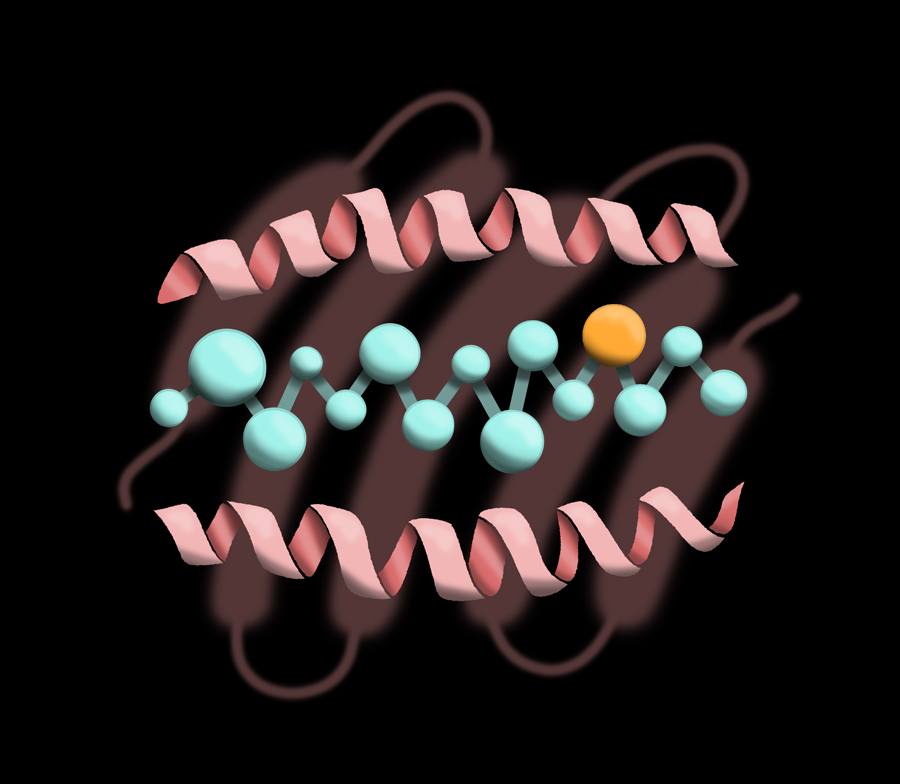
HLA-DRB1 can ‘hold peptides’ that are 15–20 amino acids long and present them to T cells. These illustrations depict the amino acid peptide sitting in the pocket of HLA-DRB1.
The team think that when both HLA-DRB1*16:02 and a copy of the ancient mutation are present, the allele peptides boost immune system self-recognition, so antibodies are not produced against the body’s own proteins. But here is the surprising bit – this combination was protective only in more obese individuals. This finding is counterintuitive because obesity usually increases the risk for T2D. Conversely, the SLC16A11 mutation is associated with higher risk of T2D in leaner individuals. This switch from risk to protective is the opposite of expected and presents a perplexing conundrum that needs further study.
Looking ahead
The study improves our understanding of which genes, specifically HLA alleles, contribute to T2D or protect against it. It highlights that this area of study is even more complex than previously thought. The study sheds light on why Indigenous Americans have such a high prevalence of T2D but also helps explain why some people do not develop the disease. With immune genes now implicated in the development of T2D, it seems autoimmune pathology is not exclusive to Type 1 diabetes.

But here is the surprising bit – this combination was protective only in more obese individuals. This finding is counterintuitive because obesity usually increases the risk for T2D.
The surprise finding of a switch from risk to protection and its dependence on BMI raises questions that need answering in future studies, with Williams and his colleagues at the forefront of this rapidly growing field of research. His recently published book offers a personal account of his early life that provided the foundation for an extensive career in biological anthropology and human genetics. In these memoirs, Midcentury tales from rural Ohio: Birth, growth, and near death, Williams discusses his childhood and life in Ohio and lessons from his scientific career.
What led you to study genetics and genetic epidemiology of Indigenous Americans?
The primary factor was serendipity, by chance being in the right place at the right time. In 1970, when I came out of The United States Marine Corps and went back to finish my undergraduate degree at The University of Michigan, I decided, after previously being a chemistry major, to change my concentration to biological anthropology because I had taken anthropology courses and was attracted to the subject of human variation. For me, human genetic variation was the key, and I directed my courses and studies to learn all that I could. I was fortunate that a member of the Michigan faculty was Professor Frank Livingstone who participated in the early description of the sickle cell polymorphism and its interaction with malaria in Africa. Livingstone was a wonderful teacher of anthropological genetics, and became my PhD mentor and advisor.
After graduation and a year’s graduate work at Michigan, I was awarded the Eugene Power Exchange Scholarship to Magdalene College Cambridge where I was to pursue a Part 2 Degree in Biological Anthropology. There I met the Director of the HLA laboratory at Old Addenbrookes Hospital who agreed to take me on as a research student in my second year. I learned all about the new emerging field of HLA serology, genetics, and immunology and completed a research project that I put toward completing my Cambridge BA degree. It was later published in a British medical journal. Arriving back at Michigan, I used the project as the basis for my PhD research. In 1976, I was awarded a two-year NIH Postdoctoral Research Training Fellowship in the Hospital’s Department of Human Genetics with Drs James Neel and Henry Gershowitz, who was a pioneer and expert in the Gamma Marker (Gm) IgG genetic system. I joined Dr Neel’s Trio Study and looked at mother-foetal immune interactions using the Gm system as a probe.
In the fall of 1978, when I arrived to take up my position as a tenure-track Assistant Professor at Arizona State University, I became aware of the long-range study of Indigenous Americans in Arizona that was begun by the NIH Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) in 1965, and made an appointment to talk to the head of the epidemiology section. Here is where the serendipity comes in: the study had been collecting blood samples and typing members of the Indigenous community for years, under strict rules of procedure and informed consent. He and his colleagues had a computer full of HLA, Gm, and red blood cell genotypes but little understanding of their serology and genetics. We published our first HLA-diabetes paper in 1981, and the first genetic evidence of a three-migration model for Paleo-Indians across the Bering Strait in 1985 using a large sample of Gm data. The work has continued to this day with our latest collaboration highlighted here.
What is one common misconception people have about your work?
The average person thinks of a ‘race’ as a uniform collection of humans who all share the same characteristics. This is wrong. Scientists have long ago deprecated the concept. We find in the human genome great variation, within ethnic groups and between ethnic groups. We are all one variable species with no clear genetic boundary lines while genes do not come with tags; there are no white genes, or black genes, or Jewish genes, or Indian genes. There are just genes. When any two human beings conceive a child, those genes are passed on irrespective of the combination of ethnicity and religion.
How can this unexpected finding be further investigated?
Since the discovery of HLA we have been performing disease associations with two elements, the HLA molecule and the differing frequencies of the disease and HLA marker in the control and patient samples. Our paper suggests a new tack, that we need to add a third element to the studies, the peptide. This can be facilitated by creating a library of predicted and measured strong peptide binding with the proteins that have been, to date, associated with the disease.









