The Translational Genomics Group, led by Dr Ruben Artero at the University of Valencia, Spain, conducts vital molecular research to elucidate the pathogenesis of genetic diseases such as Myotonic dystrophy type 1. The group’s research into this rare disease has led to the identification of therapeutic targets such as microRNAs (miRNA) and the development of innovative therapies (antagomiRs and blockmiRs) against these targets. This significant preclinical research provides a platform for further studies to help address the unmet need for effective treatments. Through the team’s spin-off company, ARTHEx Biotech, they are also working to overcome resource limitations, contributing to a new era of RNA-based drugs.
Myotonic dystrophy type 1 (DM1) is a rare, genetic, multisystem progressive disorder, with a recent estimated prevalence of 9.27 per 100,000 people, worldwide. Symptoms include muscle weakness and wasting, delayed muscle relaxation after contraction (myotonia), gastrointestinal symptoms and neurological impairment. These symptoms are highly variable between patients, and serious or even fatal manifestations include heart defects and respiratory failure. No effective treatment exists and there is a need to develop therapies that relieve symptoms and improve the quality of life of DM1 patients.
Translational research aims to translate scientific findings into real life benefits for patients. It is vital in order to expedite basic scientific discoveries into practical clinical treatments for, or prevention of, diseases. Using human cells and animal models, the Translational Genomics Group led by Dr Ruben Artero at the University of Valencia, Spain, works to gain a deep understanding of the complex molecular pathogenesis of DM1. In doing so, their valuable translational research enables therapeutic candidates to be identified and potential therapies to be evaluated.
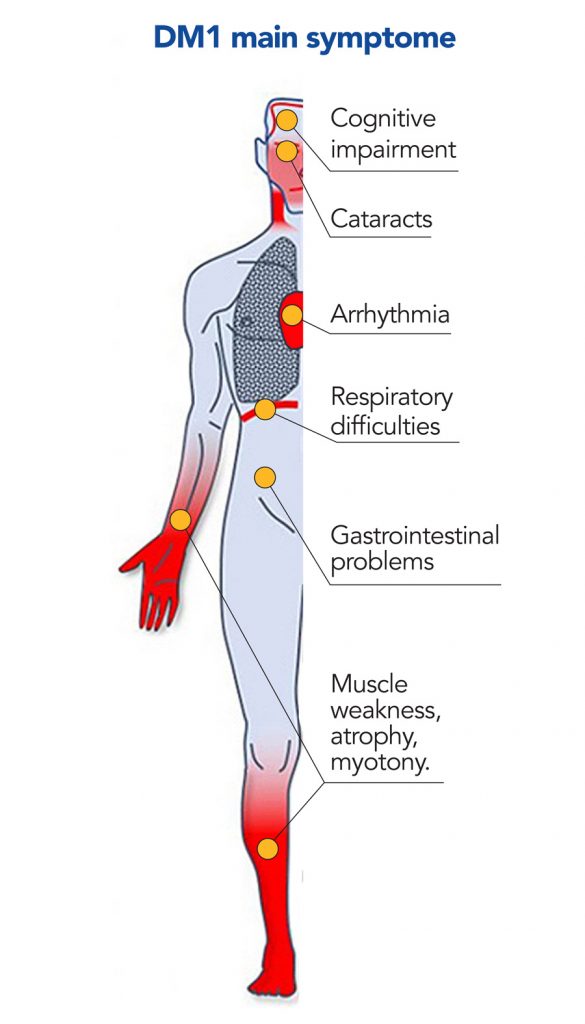
DM1
This genetic disorder stems from an abnormal repetition of the trinucleotide CTG sequence in a non-coding region of the DM1 protein kinase (DMPK) gene. These long CTG sequence repeats cause the accumulation of toxic RNA transcripts in the cell nucleus of skeletal muscle and, to a lesser extent, in cardiac and brain tissue. This toxic RNA sequesters the muscleblind-like family of proteins (MBNL) which are essential for RNA metabolism and gene expression functions. MBNL proteins have fundamental roles in alternative RNA splicing and the initiation of adult RNA splicing patterns. Precursor mRNA forms mature mRNA that is translated into proteins, therefore if precursor mRNA is altered, this can affect the expression level and functionality of proteins.
The vast majority of mRNA in our cells undergo alternative splicing (the process whereby different combinations of splice sites are selected within a messenger RNA precursor), highlighting the importance of this process in health and disease. Depleted MBNL protein levels cause irregular alternative RNA splicing of hundreds of genes. In addition to the direct binding of toxic RNA to MBNL proteins, toxic RNA also activates antagonists of MBNL, namely CUGBP elav-like family member 1 (CELF1) protein. Such activation results in a gain of function of CELF1, a protein involved in RNA processes including the promotion of foetal patterns of alternative splicing, which results in dysfunctional proteins and aberrant physiological processes manifesting in a broad variety of symptoms. The pathogenesis of DM1 also includes the disruption of several signalling pathways important for normal cell physiology.
“Although defined as a rare disease, globally, DM1 is debilitating for hundreds of thousands of people and there is a lack of effective treatment.”
Overview of DM1 therapy strategies
Given that the loss of function of MBNL is a pivotal aspect of the pathophysiology of DM1, MBNL is a key target for therapy. One approach is to target the toxic RNA which inhibits MBNL protein function. Other approaches include trying to increase the amount of MBNL or its activity in cells while trying to regulate CEFL1 levels. Three main categories of therapies are being explored to address this.
The first category are ‘small molecules’ which are often repurposed drugs already used in other diseases and with proven safety in humans. One example in phase II trials is Tideglusib, a regulator of CELF1 levels in DM1 leading to improvements in symptoms in childhood-onset DM1.

dystrophy type 1, and the difference between amtimiRs and blockmiRs ways of action.
The next category are oligonucleotide-based therapies, consisting of short sequences of RNA or DNA with complementary or antisense sequences against a target RNA. The mechanisms of action of antisense oligonucleotide (ASO) include targeting abnormal DMPK transcripts and preventing pathogenic sequence repeats from inhibiting MBNL1. One of the limitations of the ASO treatments is the insufficient delivery to certain tissues and conjugation to delivery systems is under investigation to address this.
Gene therapies are the third therapeutic category. Therapies to increase/decrease the expression of genes and proteins offer a viable option and warrant further research. Two approaches are commonly adopted: the use of viral vectors to deliver a gene that can be used to produce a protein, and CRISPR/Cas9 systems to edit the mutated gene.
There are numerous other drugs under investigation for their potential benefits in treating clinical manifestations of DM1 – particularly manifestations such as pain, myotonia, and daytime sleepiness – but they do not target the origin of the disease.
Micro RNAs as therapeutic targets
Micro RNAs (miRNA) are small non-coding RNAs which regulate mRNA levels and function by inducing their breakdown and affecting mRNA translation into proteins. They have been implicated in other diseases but are now being studied as potential therapeutic targets in DM1. In DM1, studies reveal there is an alteration to the miRNome, and numerous miRNAs are dysregulated. The Translational Genomics group has shown that the expression of DMPK toxic RNA increases the levels of miR-218, which repress the translation of MBNL proteins and reduce its levels. By targeting miRNAs using ASOs engineered to be complementary to them (antagomiRs and blockmiRs), it is possible to restore MBNL function. They act to ‘silence’ miR-23b and miR-218, and in a study of human DM1 myotubes and a mouse model, the group demonstrated that treatment with these antagomiRs increased MBNL levels and prevented irregular RNA splicing, improved myotonia and other pathological aspects of DM1.
Different studies by the group published in Molecular Therapy: Nucleic Acids investigated antagomiR-23b therapy and uptake of antagomiR-218 in mice, demonstrating low toxicity and effective tissue delivery. Dose-dependent increases in MBNL protein levels and improvements in grip strength and myotonia were found. Effects started to diminish four days after injection but reduced myotonia and improved grip strength benefits continued 45 days post treatment. The study confirmed the true potential of this treatment for DM1 patients, and further research is ongoing.
“To progress this area of research, improved preclinical models and drug evaluation methods are much needed.”
We are now witnessing the advent of a new era of RNA-based drugs and vaccines. Compared to other chemical drugs, ASOs bind a very specific sequence, meaning they act in a highly targeted manner. Once a therapy target has been identified, antisense molecules can be used to inhibit the development of a disease. In the case of muscle tissue, they do not require complex delivery systems to get to the cells and block these miRNAs; when molecules are tiny enough, a simple injection can be sufficient.
ARTHEx Biotech and the TATAMI Project
The importance of translational research is undisputed, and without it, vital discoveries will not be implemented into practical solutions as quickly and efficiently as required. However, several challenges exist and limited funding and resources are key obstacles faced by researchers. To generate the much-needed therapies for DM1 patients, close collaboration between academics, clinicians and pharmaceutical developers is needed. Initiating clinical trials requires extensive resources, documentation, and knowledge in order to gain approval and to enrol a sufficient number of patients, especially for a rare disease such as DM1. Ongoing monitoring and management are also required as well as sufficient funds for new drug development.
The group’s translational strategy strives to overcome these challenges through multidisciplinary academic and industrial research which draws on a mixed model of public and private investment. Such collaborations are needed to facilitate funding and pool the required skills and knowledge to address the aforementioned challenges.

A successful example of a collaboration between researchers and public and private funders is the group’s spin-off company, ARTHEx Biotech, and the TATAMI project consortium, where all the participant institutions are public, but the funding is private. Both initiatives aim to explore and develop candidate ASO therapies for DM1.
ARTHEx Biotech investigates novel therapies for microRNAs manipulation in DM1, and is an excellent example of translating scientific findings into potential therapies. TATAMI (Therapeutic targeting of MBNL microRNAs as innovative treatments for myotonic dystrophy) is a project funded by La Caixa Foundation and carried out by an international consortium led by Artero’s Translational Genomics Group. Building on the team’s findings related to the beneficial effects of antagomiR-23b and antagomiR-218 therapies in DM1 mice models, the group explore and test numerous ASO variants. These antagomiRs undergo comprehensive in vitro testing and optimisation to identify the most suitable candidates which are further analysed in vivo in mouse models, before final characterisation in human cells. Following this project, resources and funding to take these findings into clinical trials is required so that a new RNA-based drug for the treatment of DM1 can hopefully be implemented. Such therapies could be further explored for use in other diseases.
To progress in this area of research, improved preclinical models and drug evaluation methods are urgently required. Moving beyond 2D culture methods to employ tissue engineering to create 3D muscle models which more closely represent in vivo conditions is an area of interest. The group has collaborated with TATAMI partner IBEC to develop an in vitro 3D DM1 model of human skeletal muscle to be used in preclinical studies of DM1. Such models aim to further enhance our understanding of DM1 pathogenesis and facilitate evaluations of drug delivery and activity.
Although defined as a rare disease, DM1 is debilitating for hundreds of thousands of people and there is a lack of effective treatment. Overall, these innovative miRNA therapeutic targets are in the preclinical phase but offer great promise to be tested in the medium term in humans. With the rapid growth in our ability to obtain a wealth of genomic and proteomic data, it is hoped that further therapeutic targets will be discovered and effective clinical treatments developed. The Translational Genomics Group aims to be at the forefront of these RNA-based drug developments for the treatment of DM1, through ‘bench to bedside’ translational research.
As researchers, there are many challenges to face when investigating rare diseases, and the scarcity of patients to study is only the most obvious. However, the most worrying is that the translation of drug candidates from the lab bench to the bedside of patients is an extremely difficult process. Institutions lack appropriate support structures, and clinicians and researchers involved often lack proper training to engage the industry. It is necessary to improve the implication and cooperation of the different actors to achieve adequate treatments against diseases such as DM1.
References
- Overby, SJ, Cerro-Herreros, E, González-Martínez, I, et al, (2022) Proof of concept of peptide-linked blockmiR-induced MBNL functional rescue in myotonic dystrophy type 1 mouse model. Molecular Therapy: Nucleic Acids, 27, 1146–1155. doi.org/10.1016/j.omtn.2022.02.003
- Pascual-Gilabert, M, López-Castel, A, Artero, R, (2021) Myotonic dystrophy type 1 drug development: A pipeline toward the market. Drug Discovery Today, 26(7), 1765–1772. doi.org/10.1016/j.drudis.2021.03.024
- Cerro-Herreros, E, González-Martínez, I, Moreno, N, et al, (2021) Preclinical characterization of antagomiR-218 as a potential treatment for myotonic dystrophy. Molecular Therapy: Nucleic Acids, 26, 174–191. doi.org/10.1016/j.omtn.2021.07.017
- Fernández-Garibay, X, Ortega, MA, Cerro-Herreros, E, et al, (2021) Bioengineered in vitro 3D model of myotonic dystrophy type 1 human skeletal muscle. Biofabrication, 13(3), 035035. doi.org/10.1088/1758-5090/abf6ae
- Cerro-Herreros, E, González-Martínez, I, Moreno-Cervera, N, et al, (2020) Therapeutic potential of AntagomiR-23b for treating myotonic dystrophy. Molecular Therapy: Nucleic Acids, 21, 837–849. doi.org/10.1016/j.omtn.2020.07.021
- López-Castel, A, Overby, SJ, Artero, R, (2019) MicroRNA-based therapeutic perspectives in myotonic dystrophy. Int J Mol Sci, 20(22), 5600. doi.org/10.3390/ijms20225600
- Cerro-Herreros, E, Sabater-Arcis, M, Fernandez-Costa, J M, et al, (2018) miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nature Communications, 9(1), 2482. doi.org/10.1038/s41467-018-04892-4
- van den Hoogenhof, MG, Pinto, YM, Creemers, E, (2016) RNA splicing regulation and dysregulation in the heart. Circulation Research, 118(3), 454–468. doi.org/10.1161/CIRCRESAHA.115.307872
- Hahn, C, Salajegheh, MK, (2016) Myotonic disorders: A review article. Iranian Journal of Neurology, 15(1), 46–53.
- Liao, Q, Zhang, Y, He, J, Huang, K, (2022) Global prevalence of myotonic dystrophy: an updated systematic review and meta-analysis. Neuroepidemiology, [Online ahead of print: April 28, 2022] doi.org/10.1159/000524734
- Fang, FC, Casadevall, A, (2010) Editorial: Lost in Translation—Basic Science in the Era of Translational Research. American Society for Microbiology Infection and Immunity, 78(2), 563–566.
10.26904/RF-142-2906007001
Research Objectives
The Translational Genomics Group led by Dr Ruben Artero investigate innovative therapies for Myotonic dystrophy type 1.
Funding
This work is being funded by la Caixa Research Fundation TATAMI project (HR17-00268), with additional funds from the Myotonic Dystrophy Foundation (MDF), Generalitat Valenciana (PROMETEO/2020/081), Instituto de Salud Carlos III (DTS19/00128, with the ERDF).
Collaborators
Special thanks to Estefanía Cerro-Herreros and Beatriz Llamusi, and the whole Translational Genomics, ARTHEx, and TATAMI teams.
Bio

Contact
Departamento de Genética, Facultad de Biológicas,
Universidad de Valencia
Dr Moliner, 50 / 46100 Burjasot, Valencia, Spain
![]()
E: ruben.artero@uv.es
T: +34 963543028