
ALCAPA is a rare but life-threatening heart disease which leads to life-long and life-threatening arrhythmia. As a congenital disorder, one that develops in the foetus, research has mostoly focused on infant ALCAPA cases, at the expense of adult cases. Yet nearly 20% of adult cases lead to a sudden death upon its development, and as non-invasive detection technology has improved, more cases of ALCAPA in adults have been detected. Using histology samples from adults with ALCAPA, Dr Hiroshi Kubota from Kyorin University set out to learn about ALCAPA in adults and meet this crucial research need.
Anomalous left coronary artery from the pulmonary artery (ALCAPA), also known as Bland-White-Garland syndrome, is a heart disease which affects how the heart muscle itself receives blood. Stripped to its most fundamental purpose, the heart is another muscle, albeit with some unique features. Thus, even the heart needs the steady flow of blood to deliver oxygen to its cells and dispose of carbon dioxide to pump blood around the body.
The heart wants what the heart wants
The blood circulation to the heart itself, known as coronary circulation, is arguably the most important circulation system in the body. Oxygenated blood is supplied by coronary arteries, which are divided into two main pathways, the left and right coronary arteries, which are roughly responsible for distributing oxygenated blood to their eponymous sides. They continue to divide into marginal arteries to supply the whole heart with blood. These arteries are intricately wrapped around the heart muscle.

People who develop ALCAPA, however, are unable to receive any oxygenated blood through coronary circulation. A typical coronary circulatory system receives its blood from the aorta – the artery that the heart pumps oxygenated blood out to the rest of the body. But the left coronary artery (LCA) in a person with ALCAPA is miswired, and receives its blood from the pulmonary artery, which is the artery that carries deoxygenated blood to the lungs for its re-oxygenation.
What that means is that the blood supplied from the pulmonary artery, and through the left coronary artery, has a very low concentration of oxygen. Imagine if the taps of water in your house were receiving your wastewater from your toilet and shower. The very water that should be leaving your house for treatment to re-enter the water cycle is being funnelled back into your home by bad plumbing.
“Aside from basic information and the established treatment protocol for patients, not much is known about ALCAPA in adults.”
This is as catastrophic for the heart as it would be for your home. Many heart attacks and diseases arise due to complications with coronary circulation, as the heart muscle slowly weakens from a lack of oxygen. The ultimate consequence are heart attacks and heart failures for the oxygen-starved heart muscle. Unsurprisingly, ALCAPA is a very serious disease that must be treated immediately.
What is ALCAPA?
Despite the severity of ALCAPA, you’d be forgiven for answering that question as it’s a rare disease. ALCAPA is only estimated to occur in 1 of every 300,000 births.
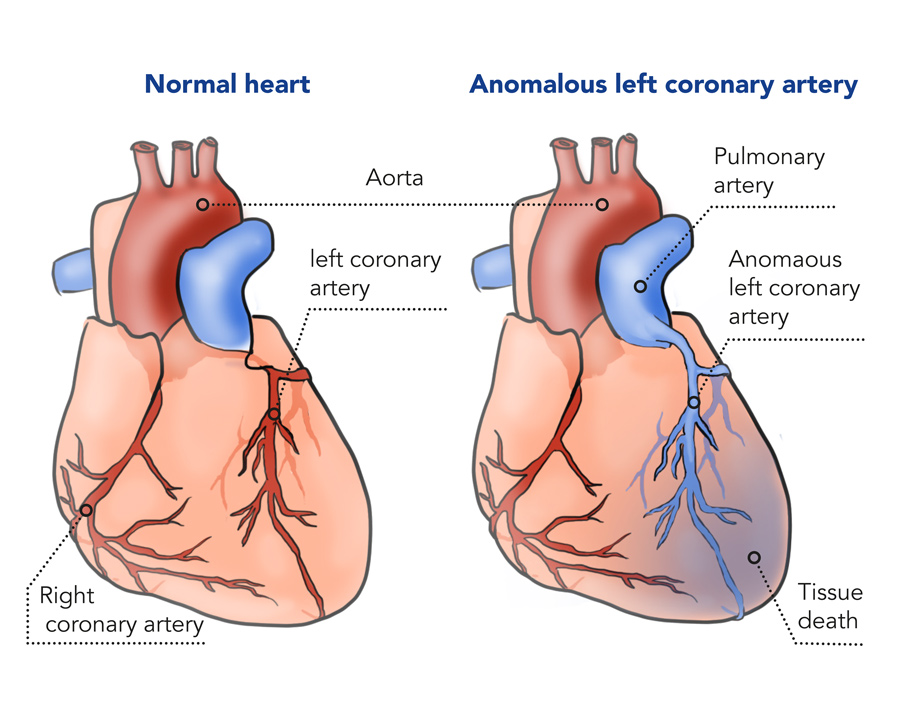
The disease is typically associated with infants as a congenital disease – a disease that arises in the development of the foetus within the womb. ALCAPA begins as an asymptomatic disease, but symptoms like rapid breathing and bad feeding will arise within a few months due to a drop in pulmonary blood pressure. Any babies identified with ALCAPA symptoms are rushed into emergency surgery. Though surgery isn’t ideal, its preferable to the alternative, as 9 out of 10 untreated infants will die in their first year.
Most newborn patients with ALCAPA syndrome die from global ischemic cardiomyopathy. This means that the muscle in the chambers of the heart are stiffened due to less blood supply from the coronary arteries, eventually leading to death from severe ischemic heart failure. However, a few patients can survive to adulthood if they develop adequate collateral circulation via a blood vessel between the right and left coronary arteries which can pump blood from the right coronary artery to the left coronary artery.
Cases of untreated ALCAPA have been found in adults. As our ability to diagnose ALCAPA without invasive and somewhat messy procedures like surgery grows, academics have become more interested in ALCAPA, especially in adults.
We know a lot about the basic epidemiology and demographics underpinning ALCAPA. In a review of 151 ALCAPA cases (live cases and autopsies), the average age was 41. Still, cases in people as old as 83 years were found as well. Other autopsy studies detected that cases typically arose when people were in their 40s, with the youngest case being 35 years old.

Patients typically show symptoms of angina and short breath (dyspnea), and can show arrhythmia, with 17% of cases leading to sudden death, and approximately 15% of cases being asymptomatic. ALCAPA cases were also seen to disproportionately impact women by a 2:1 ratio.
Aside from this basic information and the established treatment protocol for patients (surgery as soon as possible), not much is known about ALCAPA in adults. This is because research has focused predominantly on infant ALCAPA cases. As a result, there are many open questions about its impact on the body. How does the body respond to surgery? How effectively does the body heal? What are the long-term health impacts?
Learning about ALCAPA in adults
Dr Hiroshi Kubota, a cardiovascular surgeon at Kyorin University, wanted to answer these questions and address this significant gap in our knowledge. As cases are rare, it’s difficult to conduct the wide-ranging, systematic analyses and use hundreds of cases to develop randomised controlled trials, which are popular and reliable methods in medical science. But Dr Kubota and his colleagues were able to treat two adult ALCAPA patients.
These two patients were rescued from ventricular fibrillation – when the heart is unable to pump blood due to erratic heart beats which causes unstable and high rates of contractions, up to 500 beats per minute (for reference, the average beats per minute for a healthy adult is between 60 and 100). Both patients were operated upon as their treatment. One survived without any problems, living the next decade with no complications, whilst the other patient had another operation nine months after their first operation, and eventually died.
“Dr Kubota found there were stark differences in the patient’s coronary circulation, both before and after their operations.”
Dr Kubota wanted to find out if there were any long-term problems, both before surgical intervention and after an operation, associated with inserting a dual coronary artery system into the patient to stop ALCAPA. One paper did look at the impact of reduced blood flow in the left coronary artery in pigs without operations, in an attempt to understand the impact on the coronary circulation system. The researchers found that the coronary artery system had undergone remodelling, with the walls of the arteries thickening, decreasing its diameter and the volume of blood pumped towards the heart.

Previous research assumed that the structural problems in coronary circulation would be corrected and repaired after the operation. This was problematic, however, as no previous research had been conducted to analyse patients after receiving corrective ALCAPA surgery, and their body’s response towards it.
Dr Kubota found there were stark differences in the patient’s coronary circulation, both before and after their operations. Looking at samples of their tissue under a microscope, they found that the arteriolar walls had thickened as well, decreasing its diameter, thus restricting its blood flow.
They also noted there were signs of “patchy fibrosis” in the heart tissue after the operation. Fibrosis generally refers to improper healing of tissues in the body, leading to tissue scarring. The scars mean the organs no longer work as effectively.
In specific reference to the heart, this is common when the heart has been damaged from a heart attack and some parts of the heart muscle experience fibrosis as a way to heal a broken heart after the initial heart attack. But it makes the heart muscle stiffer, and less effective as a pump for blood. Its development is associated with heart failure.
Orrathai Poolsawat/Shutterstock.com
Looking to the future of ALCAPA research
Data from the samples showed that, including the aforementioned symptoms, there were numerous physiological changes associated with coronary heart disease. Even after the operation, both patients were still more susceptible towards heart problems. Despite these problems, however, the surgery itself is effective. Part of the surgery included adding a defibrillator which could stabilise the patient’s heartbeat if signs of arrhythmia show. But, at least in the surviving patient, the authors didn’t detect any instances of its activation, showing that the surgery helped treat ALCAPA in adults.
Developments in technology for resuscitation have made us more aware of ALCAPA and able to prevent more ALCAPA-related deaths. Dr Kubota suggests that we look at some new medication, either vasodilators (which make the artery and veins wider), or anti-arrhythmic drugs (which stabilise the heart’s rhythm and make it less erratic).
However, there is still a clear need to conduct new research to better understand ALCAPA, particularly in adults, who have traditionally been ignored in ALCAPA research. Through this, we create new and better protocols to treat and tackle this heart disease.
Why do adults who were asymptomatic of ALCAPA at birth suddenly start showing symptoms and developing coronary heart problems?
That’s a very important question. About two thirds of adult ALCAPA patients had symptoms of angina, dyspnea, palpitations, or fatigue and about 62% of patients with life-threatening ALCAPA did not show any antecedent symptoms. My hypothesis is that both the thickening of the arteriolar wall and patchy fibrosis develop very slowly, so that patients with severe disease show symptoms only later in life, either suddenly or gradually.
Malignant ventricular arrhythmias can develop from an old infarct-related scar tissue or as a result of an acute ischemic event during exercise, where myocardial hypoperfusion, especially in the subendocardial region from coronary steal phenomenon may occur; hence, there is risk of sudden cardiac death. Sudden death in untreated patients with adult-type ALCAPA occurs at an average age of 35 years. Sudden cardiac death risk factors are severe left ventricular dysfunction and fibrotic changes as a possible substrate for ventricular arrhythmias. These risks develop slowly and gradually because fibrosis and ischemia due to arteriolar thickening may develop very slowly, year by year.
What future research direction would you like to see in ALCAPA?
It would be very important to collect multidisciplinary big data (narrative, electrophysiological, echographical, pathological, radiological, angiographycal, electrophysiological, surgical, medical, etc.) about adult ALCAPA with long-term follow up from all over the world. By reviewing the data systematically and examining the disease’s natural history, the effectiveness of several kinds of corrective surgery and medical treatment might be elucidated. My final aim is to be able to suggest the best order-made treatment(s) in combination with surgical/ transcatheter/medical/ICD implantation treatments for each adult ALCAPA patient.
References
- Kubota, H., Endo, Y., Ishii, H., Tsuchiya, H., Inaba, Y., et al. (2020). Adult ALCAPA: from histological picture to clinical features. Journal of Cardiothoracic Surgery, 15. Available at: https://doi.org/10.1186/s13019-020-1048-y
- Hong, H., Aksenov, S., Guan, X., et al. (2002). Remodeling of Small Intramyocardial Coronary Arteries Distal to a Severe Epicardial Coronary Artery Stenosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 22, 2059–2065. Available at: https://doi.org/10.1161/01.ATV.0000041844.54849.7E
- Robertson, S. (2018). What is Fibrosis? [online] News Medical. Available at: https://www.news-medical.net/health/What-is-Fibrosis.aspx [Accessed 17/03/2021]
- Boston Children’s Hospital (2021). Anomalous Left Coronary Artery from the Pulmonary Artery (ALCAPA). [online] Boston Children’s Hospital. Available at: https://www.childrenshospital.org/conditions-and-treatments/conditions/a/alcapa [Accessed 16/03/2021]
10.26904/RF-135-12181923651
Research Objectives
Prof Kubota aims to deepen our understanding of anomalous left coronary artery from the pulmonary artery (ALCAPA), a rare congenital coronary anomaly.
Collaborators
- Kyorin University: Hidehito Endo MD., Hikaru Ishii MD., Hiroshi Tsuchiya MD., Yusuke Inaba MD., Konomi Sakata MD., Kyoko Soejima MD., Hiroaki Shimoyamada MD., Hiroshi Kamma MD., Hayato Kawakami MD., Seiichi Taniai MD.
- University of Tokyo: Katsunari Terakawa MD.
- National Disaster Medical Center: Yu Takahashi MD.
- Tokyo Metropolitan Children’s Medical Center: Mio Noma MD.
- Jyukoukai Hospital: Takemoto Kazuya MD.
- National Center for Child Health and Development: Yukihiro Kaneko MD.
- Niigata University Graduate School of Medical and Dental Sciences: Satoru Hirono MD., Daisuke Izumi MD.,
Kazuyuki Ozaki MD. - Tohru Minamino MD.
- Jiseikai Nomura Hospital: Hideaki Yoshino MD.,
Kenichi Sudo MD.
Bio
Dr Hiroshi Kubota graduated Tsukuba University and received his medical degree from Tokyo University School of Medicine. He completed his Cardiovascular Residency at the Tokyo University, and did his fellowship at Tokyo University and Auvergne University, Clermont-Ferrand in France. He then joined the Tokyo University and Kyorin University as Assistant Professor in Cardiovascular Surgery in 1998. In 2011, he became Professor and Chairman of the Department of Cardiovascular Surgery, Kyorin University.
Dr Kubota’s research focuses on arrhythmia treatment, aortic surgery, brain protection, adult-congenital heart disease, valvular surgery, and developing new surgical ablation device. His numerous research projects have been funded by the Japanese Ministry of Education, Science and Culture; Japanese Heart Foundation; Pfizer Foundation; Fujita Memorial Medical Research Fund; Leading-edge Industry Design Project, Medical Innovation, Saitama Prefecture, Japan; and TERUMO life-science foundation.
He is an editor, associate editor, and a reviewer for numerous surgical and cardiology journals and has authored several book chapters. He also authored or coauthored numerous peer-reviewed publications.

Contact
Hiroshi Kubota
6-20-2, Shinkawa, Mitaka
Tokyo, 181-8611, Japan

E: kub@ks.kyorin-u.ac.jp
T: +81-422-47-5511
W: https://www.kyorin-u.ac.jp/English/








