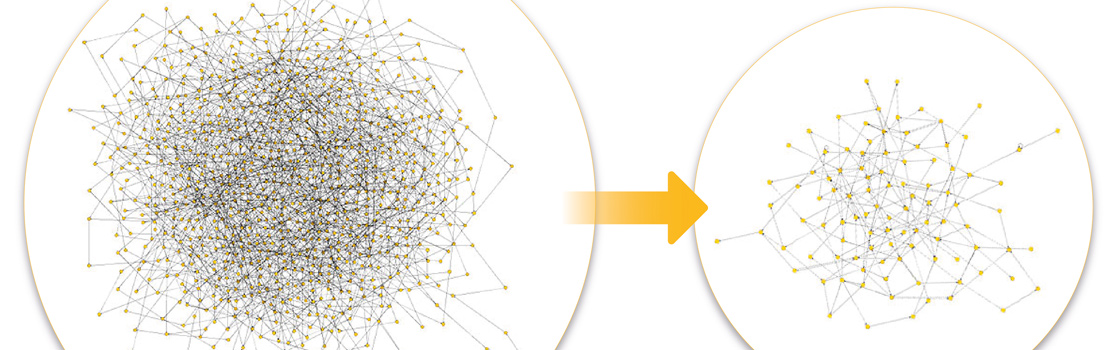
Bioengineering of organisms that produce substances useful for human consumption is a rapidly growing field. Approaches that identify key genes in bioproduction systems require unfeasible amounts of data. Dr Sachiyo Aburatani of the National Institute of Advanced Industrial Science and Technology, Japan, has applied structural equation modelling to infer causal relationships between the important genes involved in a bioproduction system. Network modelling can reveal the relationships between known and unknown genes using only gene expression data, which increases the information per unit data required to understand a biological network. Such advances can improve the overall efficiency of bioproduction.
Bioproduction is a field of bioengineering that has gathered significant pace recently, as consumer demand clashes with conservation and sustainability agendas. In Japan, the NEDO Smart Cell project aims to use bioengineering to move towards a bioeconomy, where key products in industry, healthcare, and food production are harvested from living microorganisms, such as yeasts, bacteria, and fungi. For example, reticuline – one of the key ingredients in painkillers – is produced by Escherichia coli, and the bright red pigments used in the food and meat industry are produced by Monascus fungi.
Gene network analysis
However, this process of identifying metabolic pathways and candidate genes involved in the production of a desirable compound can sometimes require unfeasible amounts of data to correctly inform the development process. Gene expression data represents the amount of gene activity or ‘product’ that a gene transcribes when it produces the proteins involved in metabolic pathways. Gene expression data can reveal some of the major genes involved in a particular metabolic pathway but cannot show the connections between genes, or show the regulatory systems involved in the entire metabolic cycle. Gene network analysis can show the interactions between genes, making efficient use of a limited dataset.

Structural equation modelling
Currently, gene regulatory networks are deduced using sophisticated statistical techniques to infer the genes related with several types of biological systems from the microarray or RNA sequence techniques as gene expression data. However, conventional statistical methods only infer direct causal links and do not suggest other candidate factors that might be related in the regulatory network. Structural equation modelling (SEM) explicitly addresses this gap and provides direct causal links as well as suggesting latent factors, or unknown variables, for further exploration. Structural equation modelling also tackles the problem of unknown data, because it can infer protein expression regulations from gene expression data and shortcut the process of gathering the large amounts of data necessary for alternative approaches such as machine learning.
“Gene network analysis can show the interactions between genes making efficient use of a limited dataset.”
Another aspect of statistical modelling is selecting the best fit to the data based on the available information. Model selection is often performed using set criteria of thresholds for including or excluding variables within the model, to produce the best fit to the data, while retaining enough generality to predict realistic, and perhaps undiscovered, patterns of biological pathways.
Network modelling
Dr Sachiyo Aburatani, from Japan’s National Institute of Advanced Industrial Science and Technology (AIST), has developed a unique algorithm to apply SEM to estimate the optimal network model structure. Her algorithm has helped to improve model fit (by reducing the amount of unknown variance) and consistency (by reducing the sensitivity of the network to changes). Essentially, this is a way of automating the process of structural equation model formation and parameter selection, whereby parameters that are deemed to be statistically significant – that is, have a very small likelihood of the outcome occurring due to chance – are retained, while those not statistically influential are removed. This process is reiterated, changing one parameter at a time, until the network retains only those connections and nodes that are deemed to be of significance. The process also works in reverse, as the modification index (MI) scores can be used to input new parameters that significantly change the fit of the network model. At each stage of the iterative process, the model is redrawn and re-evaluated until an optimum version is achieved. Dr Aburatani has demonstrated that network modelling (mapping) of gene expression data using structural equation modelling can be used as a universal tool across different organisms to infer the relationships between known and unknown variables, making it a valuable addition to the bioengineering toolkit.

Gene network analysis
To illustrate its wide-ranging utility, Dr Aburatani has been applying this modified SEM technique to clarify many types of biosystems at first. Her first application was to derive the gene regulatory network model for the series of transcriptional regulation in GAL-related genes in Saccharomyces cerevisiae – commonly known as Brewer’s yeast – where she highlighted where genes were regulated by one or a combination of factors.
Dr Aburatani has also applied this approach to show the hierarchical network involved in the early embryonic development of Caenorhabditis elegans – a temperate soil nematode commonly used in research. She included transcription factor proteins as latent (unknown) variables, negating the need to collect protein transcription data. This approach identified not only key genes involved in embryogenesis, but also the regulatory pathways. Similarly, in Drosophila – a small fruit fly often found inhabiting compost heaps – Dr Aburatani discovered 35 causal relationships between 18 genes and highlighted further unknown factors involved in metabolic pathways.
Dr Aburatani’s work in mice has also examined the regulatory framework of 19 transcription factors in embryonic stem cells and showed up to four signal-transduction pathways can be involved in governing pluripotency – the fundamental basis that dictates whether a cell becomes a specific cell in the body. This highlighted the multiple, hierarchical pathways behind this important part of embryogenesis.
“Bioengineered microorganisms can also be more efficient at producing target substances than chemical synthesis in the laboratory.”
From this earlier research, Dr Aburatani has applied this SEM network modelling method to suggest the candidate genes for modification in several industrial microorganisms that produce important substances in the bioproduction industry. One application example is the oleaginous yeasts that produce triacylglyceride (TAG) – oils that are expected to be substitutes for palm oils. In this case, Dr Aburatani could identify just 14 genes are important for TAG biosynthesis in oleaginous yeast, and the gene regulatory network devised from SEM applied to 14 genes showed 27 causal relationships involved in oil accumulation. Identifying candidate genes in this way helps to narrow the search for target genes that can be modified, while including all the vital components of the bioproductive pathways.
Although not strictly in the category of bioproduction, gene network analysis can also highlight the toxic effects of chemicals on human embryos. Dr Aburatani has applied the SEM method to infer causal relationships between nine development-related genes that were exposed to 15 industrial chemicals known to affect embryogenesis in humans. The inferred networks clearly identified the potentially troubling effects that different categories of chemicals have on different developmental aspects. For example, neurotoxic chemicals, such as methylmercury, affect cell differentiation differently to neuron development. Moreover, one of the key genes involved in brain development, Oct3/4, was shown to be affected by Benzo[a] pyrene and phenobarbital, which are classified as genotoxic and carcinogenic respectively. Such applications are clearly invaluable in human health research.

Bioengineering for a sustainable future
Network modelling of gene expression data represents the whole biological system much more clearly and accurately and has the potential to identify more candidate genes than would have otherwise been found under metabolic engineering methods or machine learning approaches. This emphasis on understanding the whole cell network increases the potential for upregulating, modifying, or bioengineering more of the genes directly involved in the production of desired bioproducts. Gene network analysis can also potentially safeguard against unintended consequences, by revealing the indirect connections in bioproduct metabolism.
To date, Dr Aburatani has yet to experimentally confirm her gene regulatory networks predicted via her SEM method, and so the question remains as to how well the gene network analysis predicts the real underlying biology. While the statistical fit of the network model to known biological systems, functions, and metabolic pathways can be verified using statistical metrics and known biology, experimental confirmation is still needed for the vast number of microorganisms that are used in bioproduction. Nevertheless, the advantages of the approach in identifying candidate genes and inferring entire networks from a reduced set of observational data remain substantial.
Ultimately, the goal of a bioeconomy, whereby major industrial ingredients are produced in laboratory-maintained bioengineered microorganisms, can help to relieve some of the strain on natural resources, while meeting increasing demand from human consumers. By engineering microorganisms to produce products such as palm oil we can achieve conservation goals of reducing the spread of monocultures and reduce CO2 emissions, by replacing destructive logging with laboratory-based production. Bioengineered microorganisms can also be more efficient at producing target substances than chemical synthesis in the laboratory, further reducing the reliance on fossil fuels and making them a valued resource for all our futures.
What is your proudest discovery and why?
The most outstanding aspect of my method is that it allows structural equation modelling to be applied to biological data to estimate network structure, which was previously difficult to do. The most outstanding discovery during this project was of new candidate genes for modification in the network model, which had not been found in previous microbial breeding. The gene we discovered had no known function from the genome sequence information and could never have been discovered using conventional biological approaches. By using the network modelling technology that I have developed, the discovered genes have been shown to be involved in biological production for the first time, and I am currently in the process of obtaining a patent.
References
- Aburatani, S., Shida, Y., Ogasawara, W., et al. (2019) Application of Structural Equation Modelling for Oil Accumulation System Control in Oleaginous yeast. Journal of Physics: Conference Series, 1391, 012043. DOI 10.1088/1742-6596/1391/1/012043
- Aburatani, S. (2015) Inference of Transcriptional Network for Pluripotency in Mouse Embryonic Stem Cells. Journal of Physics: Conference Series, 574, 012138. DOI 10.1088/1742-6596/574/1/012138
- Aburatani, S., Toh, H. (2015) Network inference of AP pattern formation system in D. melanogaster by structural equation modelling. Journal of Physics: Conference Series, 490, 012145. doi.org/10.1088/1742-6596/490/1/012145
- Aburatani, S., Nagano, R., Sone, H., et al. (2013) Inference of Gene Regulatory Networks to Detect Toxicity-Specific Effects in Human Embryonic Stem Cells. International Journal on Advances in Life Sciences, 5, 103–114
- Aburatani, S. (2012) Network inference of pal-1 lineage-specific regulation in the C. elegans embryo by structural equation modelling. Bioinformation, 5, 652–657.
- Aburatani, S. (2011) Application of structure equation modelling for inferring a serial transcriptional regulation in yeast. Gene Regulation and Systems Biology, 5, 75–88. NEDO Smartcell Project www.jba.or.jp/nedo_smartcell/en/
10.26904/RF-138-1757571369
Research Objectives
Using gene expression regulatory network modelling to view the complex biological systems that take place in living cells.
Funding
- This work was supported by JSPS KAKENHI Grant Number JP24510285
- This work is based on results obtained from a project commissioned by the New Energy and Industrial Technology Development Organization (NEDO).
Collaborators
- Dr Tomohiro Tamura, National Institute of Advanced Industrial Science and Technology
- Dr Koji Ishiya, National Institute of Advanced Industrial Science and Technology
- Dr Tomokazu Shirai, RIKEN
- Dr Kazuhiro Fujimori, National Institute of Advanced Industrial Science and Technology
- Professor Wataru Ogasawara, Nagaoka University of Technology
- Dr Yosuke Shida, Nagaoka University of Technology
- Professor Hiroaki Takaku, Niigata University of Pharmacy and Applied Life Sciences
- Mr Takeaki Taniguchi, Mitsubishi Research Institute
- Mr Toshikazu Ito, Mitsubishi Research Institute
- Mr Toshitaka Kumagai, Fermlab Inc.
Bio
Sachiyo Aburatani obtained a PhD in Agricultural Science in 2003 at Kyushu University. From 2003 to 2006 she worked at the Institute of Medical Science, University of Tokyo, and since 2006 at the National Institute of Advanced Industrial Science and Technology. She is Vice Director at CBBD-OIL of AIST, and guest professor at ‘Niigata University of Pharmacy and Applied Science’ and ‘Waseda University’. She is also a guest researcher at RIKEN.

Contact
AIST Tokyo Waterfront Main Bldg, 2-3-
26 Aomi, Koto-ku, Tokyo, 135-0064,
Japan
![]()
E: s.aburatani@aist.go.jp
T: +81 3 3599 8712
W: https://www.aist.go.jp/index_en.html








